At the current time, there is no cure for amyotrophic lateral sclerosis (ALS). Things may be about to change, however. Researchers at FAU and the University of California San Diego (UCSD) have identified a protein that already displays pathological characteristics at an early stage of the neurological disease. The team has published their discovery, that could lead to a new approach for treating the disease, in the journal “Acta Neuropathologica”.
In summer 2014, amyotrophic lateral sclerosis, or ALS for short, received a lot of attention through a social media campaign. In the ice bucket challenge, millions of people across the globe emptied a bucket of ice cold water over their heads to simulate a feeling of paralysis due to the extreme cold. In Germany, approximately 6,000 to 8,000 people are living with ALS, and approximately 2,000 new cases of the disease, that proves fatal within just a few years, are diagnosed every year. “ALS is a motor neuron disease, that means it damages the nerve cells that control our muscles,” explains Prof. Dr. Beate Winner. “During the first phase, muscles become weaker, before wasting away and finally leaving patients unable to swallow or breathe independently.” The social media campaign was used to raise money for research into ALS.
Turning back the biological clock by reprogramming cells into stem cells
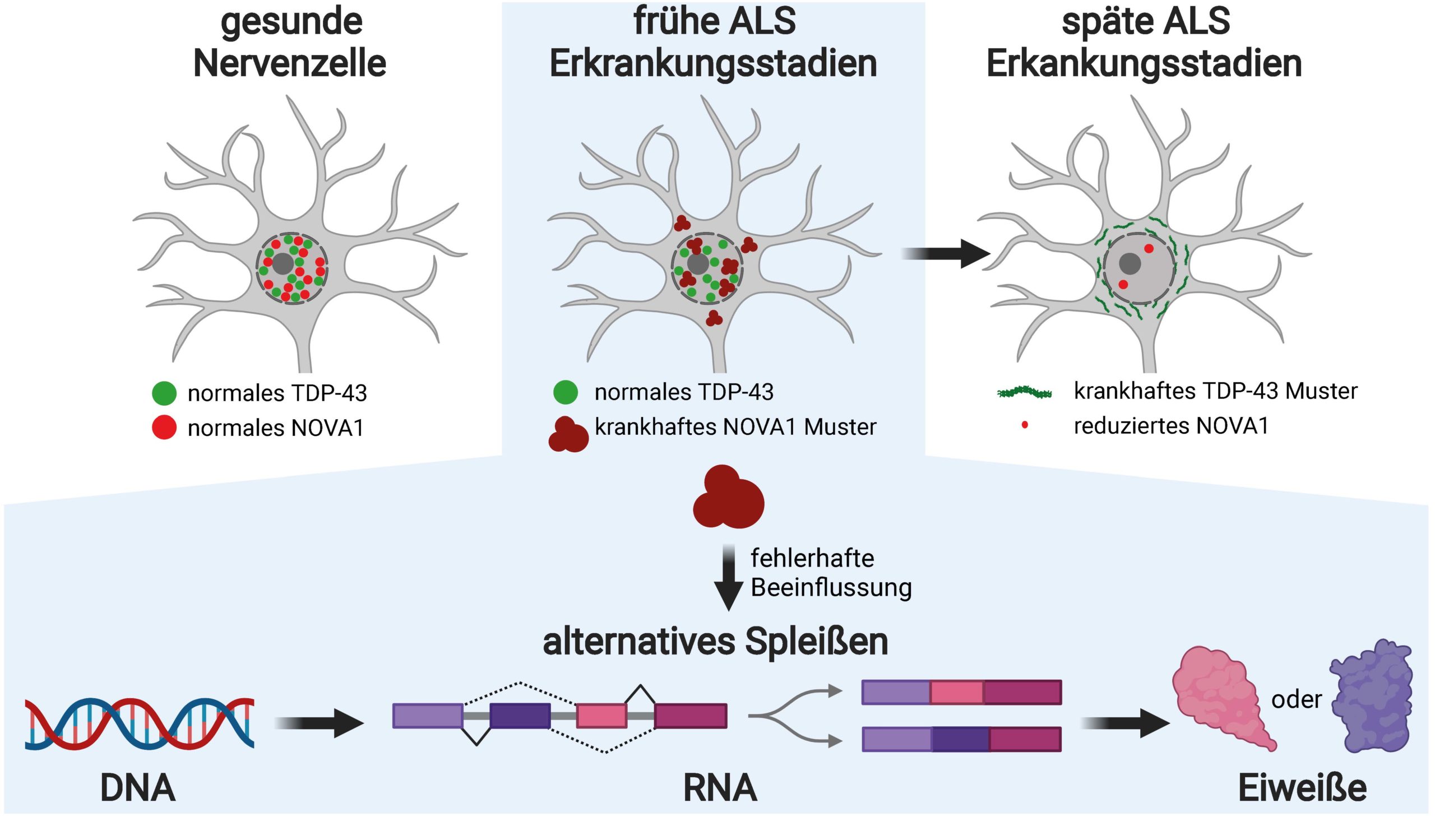
Beate Winner is a professor for stem cell models for rare neural diseases at FAU, head of the Department of Stem Cell Biology, and speaker for the Center for Rate Diseases at Universitätsklinikum Erlangen. Her laboratory investigates what triggers neurodegenerative diseases of the nervous system such as ALS in the hope of discovering new treatment options as a result. “We have known for roughly 15 years that during the end stage of ALS, the protein TDP-43 found in neurons becomes insoluble and starts to form clumps,” explains Winner. “It loses its normal functions and adopts toxic properties.” Even though these pathological changes are not yet noticeable in patients, the fate of the nerve cells is already sealed. Winner continues, “We wanted to know whether we could find causes for ALS at an early stage of development before the TDP-43 changes.”
She started her quest together with Prof. Dr. Jürgen Winkler and PD Dr. Martin Regensburger from the Department of Molecular Neurology at Universitätsklinikum Erlangen. The researchers used an innovative technique. They extracted a small skin sample from the upper arm of ALS patients and healthy people in a control group and reprogrammed it into what are known as induced pluripotent stem cells, cells that are equivalent to a very early stage of human development and that can in theory develop into any cell within the human body. These stem cells were then transformed into nerve cells. “Basically, we turned the clock back and generated neurons imitating the developmental stage of a fetus,” explains Winner. The fact that cells from adult people can be reprogrammed back into pluripotent stem cells was discovered by Shin’ya Yamanaka, who received the Nobel Prize for Medicine in recognition of his work.
The protein NOVA1 displays pathological features from an early stage
The Erlangen researchers searched for insoluble proteins in the cell samples using mass spectrometry, a high-throughput procedure. They were successful. In the nerve cells of ALS patients they discovered an RNA-binding protein named NOVA1. “In the neurons, the protein demonstrated changes including a greatly increased degree of insolvency, but not yet the typical pathological characteristics of TDP-43,” explains Dr. Florian Krach, member of the FAU team and lead author of the study. “The cells in the control group did not display these changes.”
Armed with these findings, Krach moved to the laboratory of the renowned RNA biologist and bioinformatics specialist Prof. Gene Yeo at the University of California in San Diego (USA), funded by the Bavaria California Technology Center (BaCaTeC). Thanks to specialized experiments and computer-assisted analysis he was able to investigate what NOVA1 binds to in RNA molecules and what influence it has on alternative splicing in human neurons. “Alternative splicing is an extremely complex and ingenious mechanism that humans use to multiply their repertoire of proteins,” explains Krach. “Sections of an RNA messenger molecule are either cut or added, thereby hindering, extending or changing the function of proteins altogether.”
The researchers hope their findings will help make an early diagnosis possible and open the door to new treatment concepts.
It has been known for some time that the alternative splicing process is unregulated in ALS patients. It is also known that TDP-43 influences this process. The team of researchers from Erlangen suspected, however, that other RNA-binding proteins are responsible for the pathological processes in early stages of the disease before TDP-43 changes. This suspicion has now been confirmed with the discovery of the impaired functioning of NOVA1.
“We have made a pioneering discovery, but it is only one first step towards possibly being able to detect ALS in the early stages,” says Beate Winner. “Follow-up studies with larger cohorts could deepen our understanding of the importance of RNA-binding proteins.” The researchers hope that their work will help contribute to developing new therapy concepts before neurons cross the point of no return.
Further information
DOI: https://doi.org/10.1007/s00401-022-02450-3
Prof. Dr. Beate Winner
Professorship for Stem Cell Models of Rare Neural Diseases
Phone: +49 9131 85 39301